Next: NPT Ensemble Up: Calculation of Thermodynamic Properties. Previous: Calculation of Thermodynamic Properties.
![]()
![]()
![]()
Next: NPT Ensemble
Up: Calculation of Thermodynamic Properties.
Previous: Calculation of Thermodynamic Properties.
As we pointed out in the beginning of this chapter, the main purpose of the computer simulations is to calculate thermodynamic properties. They are calculated as averages of the corresponding functions that depend on the particle coordinates. The averaging is done over the configurations, produced during the MC run.
For many quantities, like magnetization or energy, the calculations are obvious. Pressure is a little more tricky, and now we are going to derive the expression for it.
As we remember from the thermodynamics:
![]()
![]()
![]()
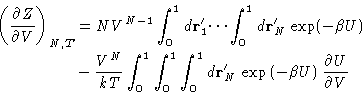 |
(1) |
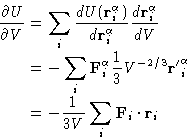 |
(2) |
Now after going back to the initial coordinates and substituting
![]() , we see,
that the first term is just density, and the second is an ensemble
average of the sum of the force times coordinate.
, we see,
that the first term is just density, and the second is an ensemble
average of the sum of the force times coordinate.
![]()
The second term on the right hand side is called virial, and this equation is often called the virial equation. If the interaction potential is pairwise, it can be rearranged as follows:
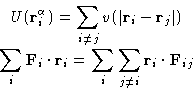
![]() is the force, acting on the atom i from the atom j.
is the force, acting on the atom i from the atom j.
![]() (Why?).
(Why?).
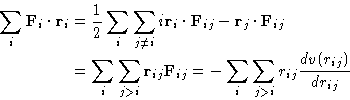 |
(3) |
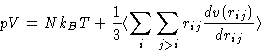 |
(4) |
If we have a system of N particles at temperature T, and volume
V, the chemical potential ![]() at large enough N can be written
as:
at large enough N can be written
as:
![]()
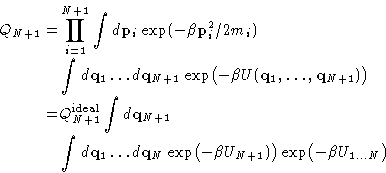 |
(5) |
![]()
The importance of the pair distribution function g(r) was discussed in the chapter on the liquid-state theory . Its calculation in the MC simulations is really straightforward. All we have to do is just to count the number of particles at the distances between r and r+dr from the chosen particle. In the homogeneous systems it is also averaged over all particles. It is calculated as a histogram with the bin width dr, and then is normalized by the area of the spherical layer.
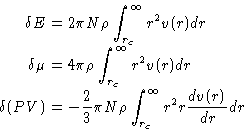 |
(6) |
© 1997 Boris Veytsman and Michael Kotelyanskii